The [Co(en)3]3+ complex is one of the classic complexes of coordination chemistry. Its stereochemistry was first correctly described by Alfred Werner in 1911.1
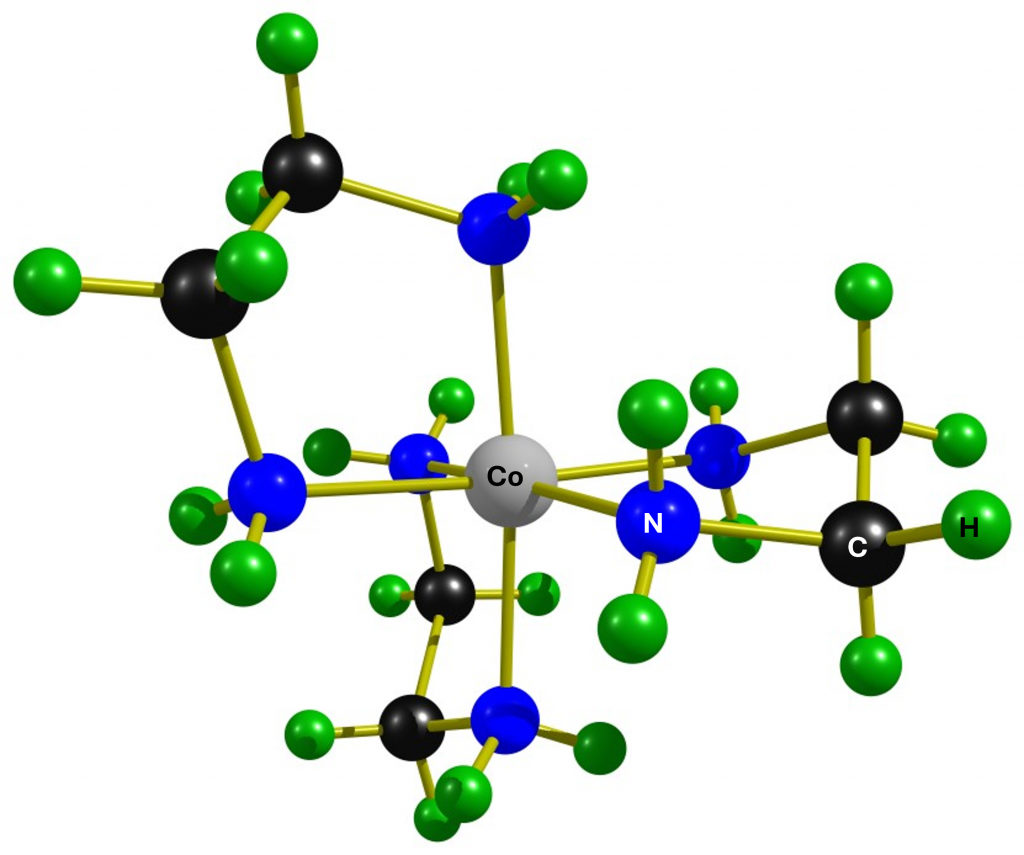
Trisethylenediamine complexes such as the cation shown in Figure 1 are always chiral. The chirality is based on the helical arrangement of the ligands (Δ or Λ isomer) and the conformation of the ligands (δ or λ), which do not form planar rings with the metal. The complexes are either D3– or C2-symmetric, depending on the conformations of the rings. The stereochemistry of such complexes is nicely explained in a detailed review article.2 The conformation of the rings plays no role in NMR spectra of this complex, as they transform into each other so quickly that the spectrometer can only measure averaged chemical shifts.
In 2024, I had the opportunity to measure a solution of [Co(en)3]Cl3 in D2O. The focus was on the 59Co spectrum, as we had not yet investigated a cobalt complex apart from the standard K3[Co(CN)6] (= 0 ppm). Since the cobalt atom in [Co(en)3]3+ is located in a very symmetrical environment, the nuclear quadrupole of 59Co should not play a significant role. Therefore, I expected a sharp, easy to measure singlet. The spectrum recorded 30 minutes after preparing the sample surprised me.
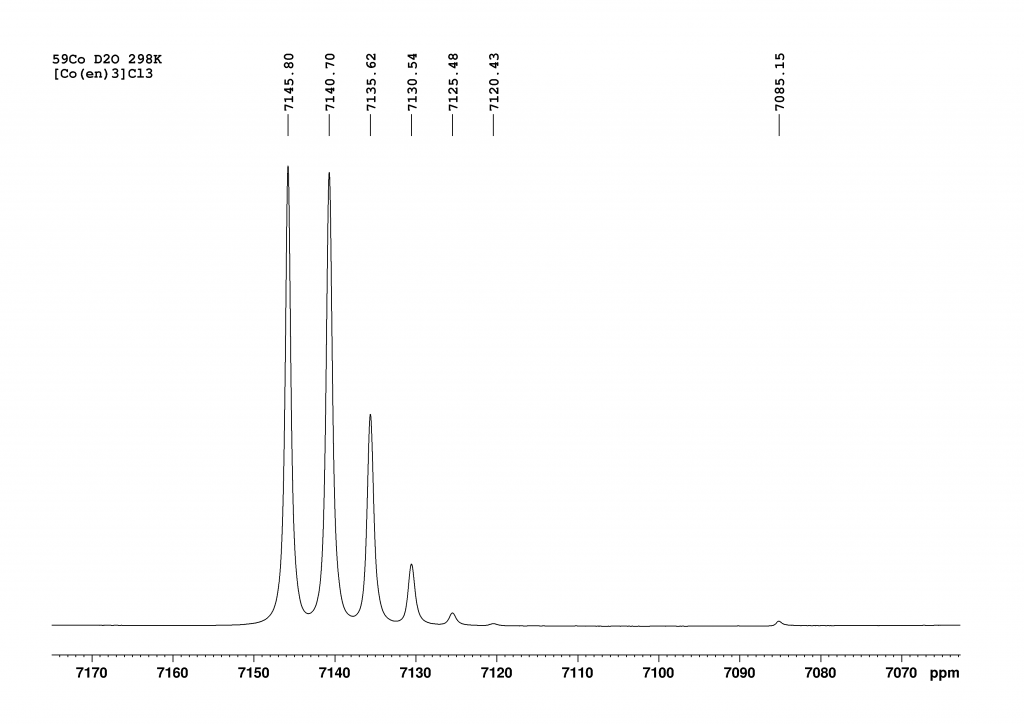
As expected, the spectrum had a very good signal to noise ratio but instead of the expected singlet, 7 peaks were visible. My first thought was that the solid we had dissolved was heavily contaminated. I was about to dispose of the sample when I noticed that the 6 peaks at low field were almost exactly 5 ppm away from their neighboring peaks. This could not be a coincidence and could not be explained by random decomposition products or impurities. However, I definitely thought the peak at high field (7085 ppm) was an impurity – a clear misjudgment.
About 24 hours after the first measurement, I wanted to look at the same sample again and repeated the measurement. This time I was really amazed by the mesmerizing spectrum I got.
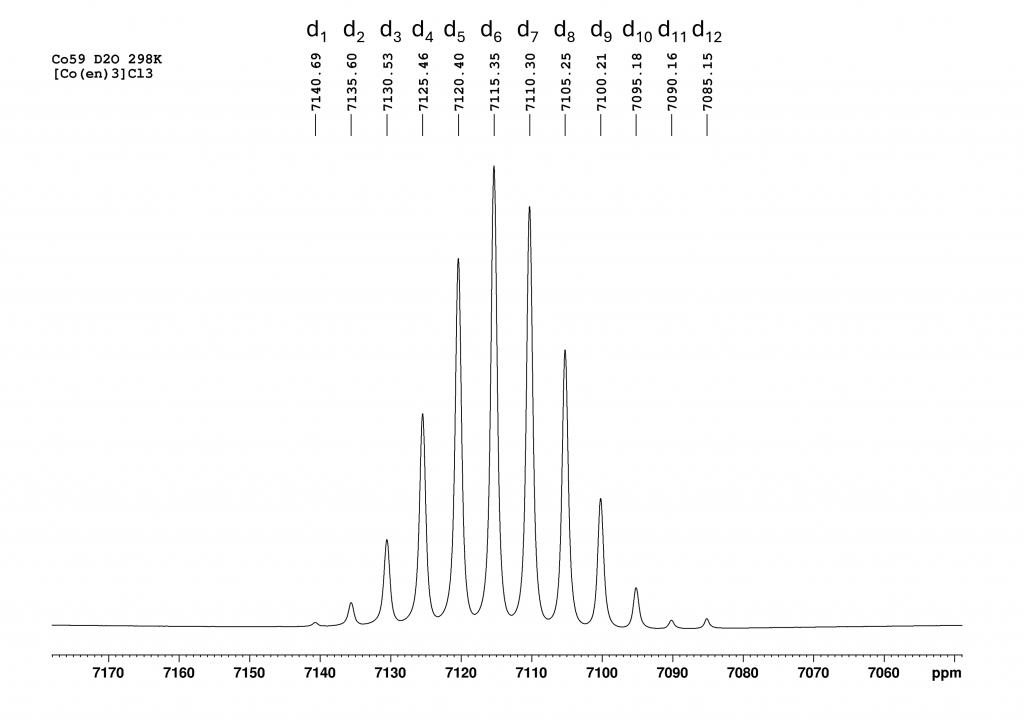
Now 12 peaks can be observed at intervals of 5 ppm and it can be seen that the signal at 7085 ppm also belongs to this ensemble of peaks. After a brief literature search, it quickly became clear what was going on here. The 12 peaks from low to high field are the isotopomers d1 to d12-[Co(en)3]3+, which were formed by H/D exchange of the NH2 protons. The signal of the non-deuterated complex (d0) has already disappeared.By integrating the CH2 protons of the ethylenediamine ligands against the remaining NH protons, the degree of deuteration can be determined at this point, which corresponds to nearby 50%. The isotope effect caused by the successive deuteration of the NH2 groups has been known since the 1980s and the kinetics of this reaction were already studied in detail at that time by Harris et al. with the aid of 59Co NMR.3 At the time, the authors only had access to a 100 MHz NMR spectrometer. The analysis of the spectra therefore had to be carried out using deconvolution. From this perspective, the kinetic analyses described are all the more impressive. In a publication by Zardrozny et al. from 2022, it was proposed to use this H/D exchange for a molecular thermometer.4
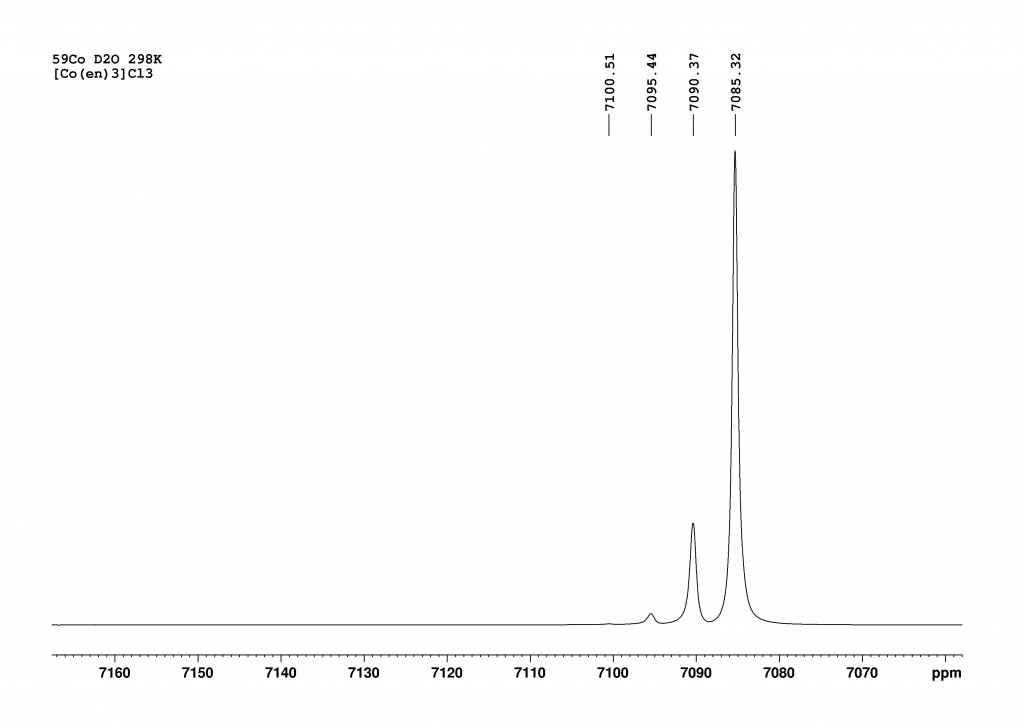
After approximately 1 week, only 4 peaks can finally be observed, with the peak at 7085 ppm (d12-complex) now dominant. The degree of deuteration is now at 98%, which agrees very well with the literature value of Harris et al. who observed the complexes d12 to d10 at equilibrium. With our much more powerful spectrometer, we also see the d9-complex at 7100.5 ppm. Due to the protons in solution, a small proportion of NH species always remains in equilibrium.
While I was working on the spectra, the question arose as to why there should only be 13 peaks (d0 to d12). In fact, there should be many more, because for most isotopomers (except d0, d1, d11 and d12) there are stereoisomers that should increase the number of peaks. For example, if we consider only the case d2, there are 3 stereoisomers (see Figure 4).
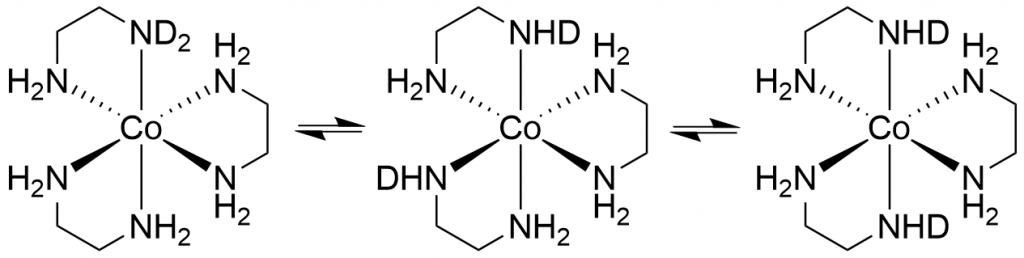
In the first there is simply an ND2 group, in the second the two NHD groups are in the cis position and in the third in the trans position. Since we only see one signal for these 3 isomers in the 59Co spectrum, I assume that there is a very fast intramolecular H/D exchange, so that the spectrometer only shows an averaged signal over the chemical shifts of the 3 isomers. This can then also be assumed for all other isotopomers. This is probably also the reason why the signals always shift by 5 ppm with each additional deuterium. Because it is not the position of the deuterium atoms but only their number that determines the chemical shift.