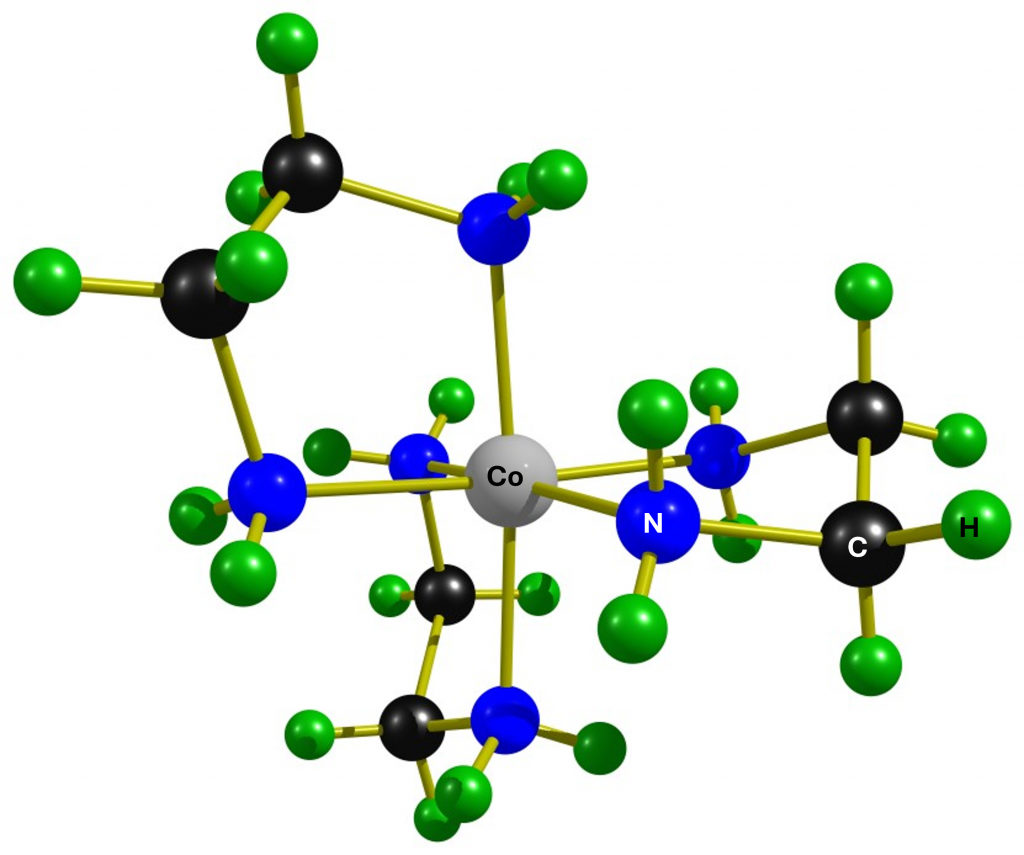
After seeing the spectacular 59CO spectra of [Co(en)3]3+in D2O,1 I thought that this could be improved upon. The impetus for this came from papers by Borer et al.2 and Iida et al.3 that described the spectroscopic differentiation of the two enantiomers of the complex using sodium tartrate and 59Co NMR spectroscopy.
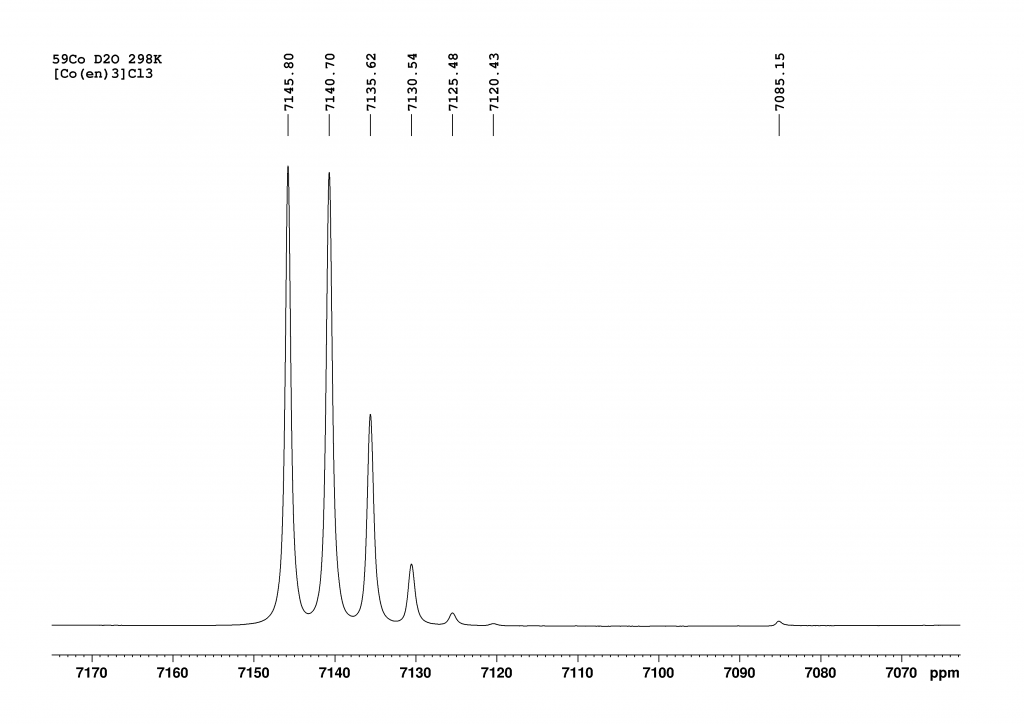
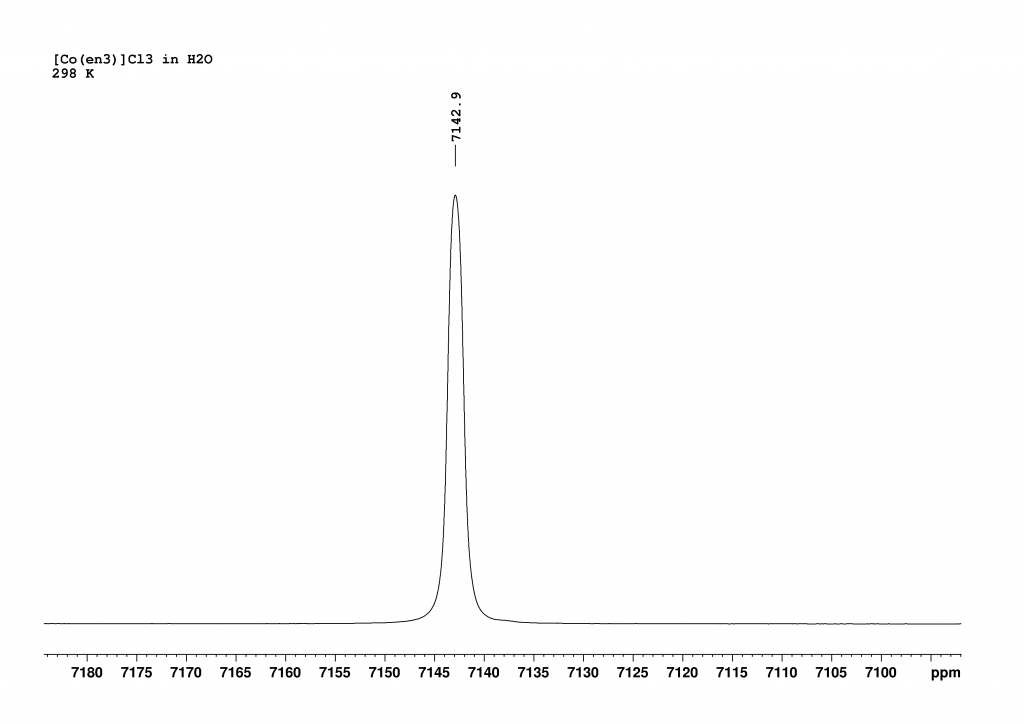
Firstly, I recorded the 59Co NMR spectrum of [Co(en)3]3+ in H2O. This revealed a single peak, which combined the signals of the two enantiomers (Δ and Λ isomers in a ratio of 1:1).
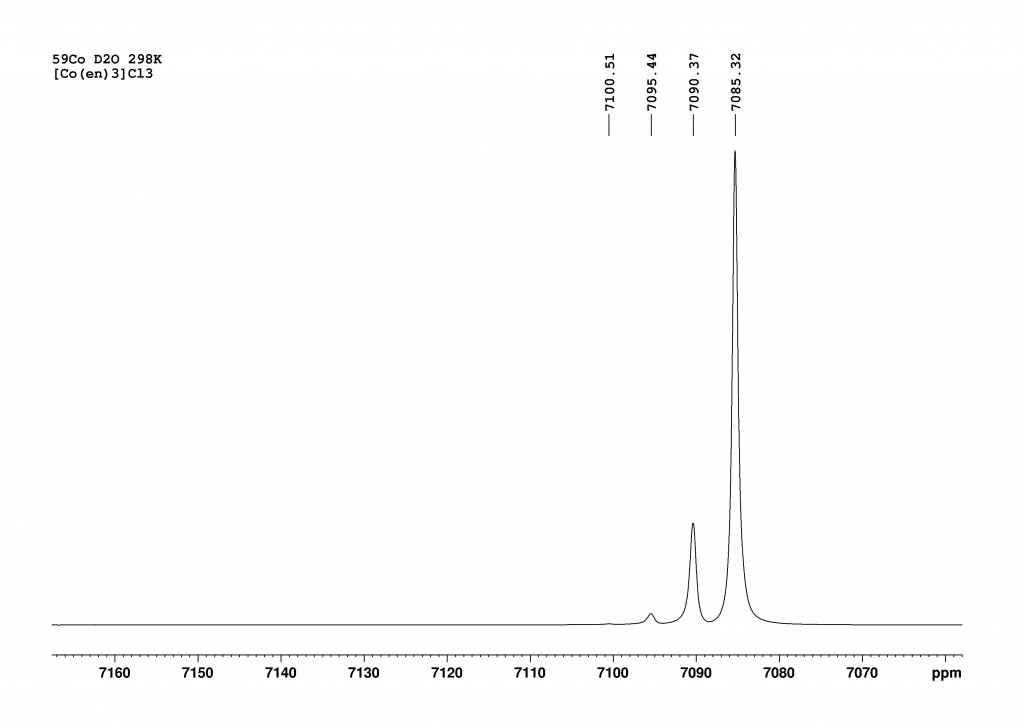
The addition of L-(+)sodium tartrate has a surprisingly clear effect. The original peak splits into two new peaks that are shifted to higher field.
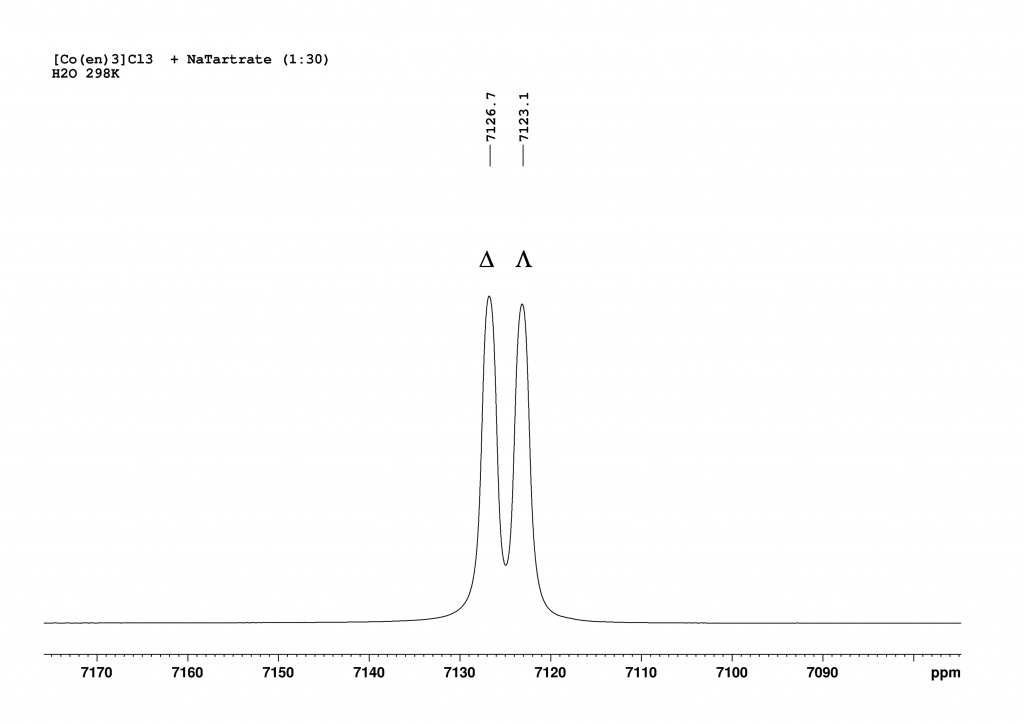
Deconvolution of the two peaks shows that they are in an integrative ratio of exactly 1:1. The cause of the splitting in solution lies in the formation of contact ion pairs, which behave like diastereomers in solution and thus result in separate 59Co signals for each enantiomeric complex.3
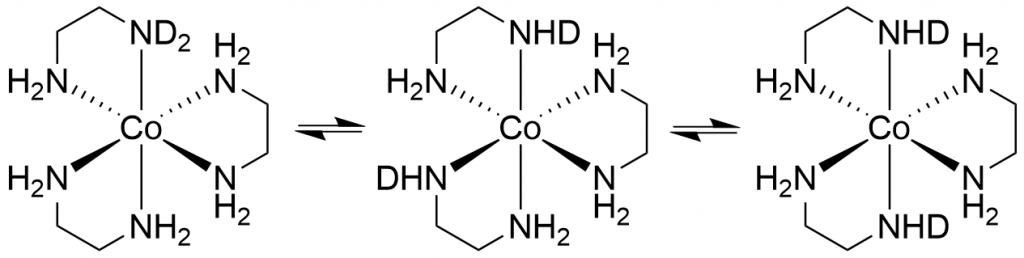
Part I of this post reported that the complex [Co(en)3]+ forms isotopomers when dissolved in D2O through H/D exchange, all of which exhibit their own 59Co NMR signal.
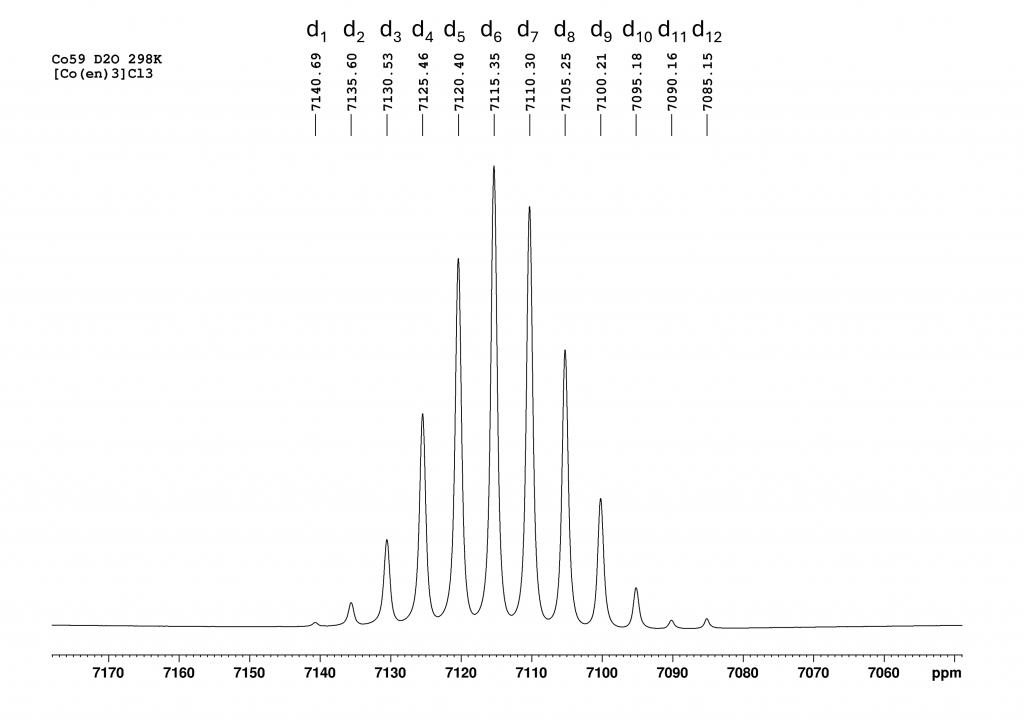
This naturally raised the question of what would happen if D2O was added to the solution of the complex in H2O with sodium tartrate. I hoped that each signal from the two enantiomers would be further split into the corresponding isotopomers. To my great delight, this is exactly what happened. The resulting spectrum is one of the most spectacular spectra I have ever measured.
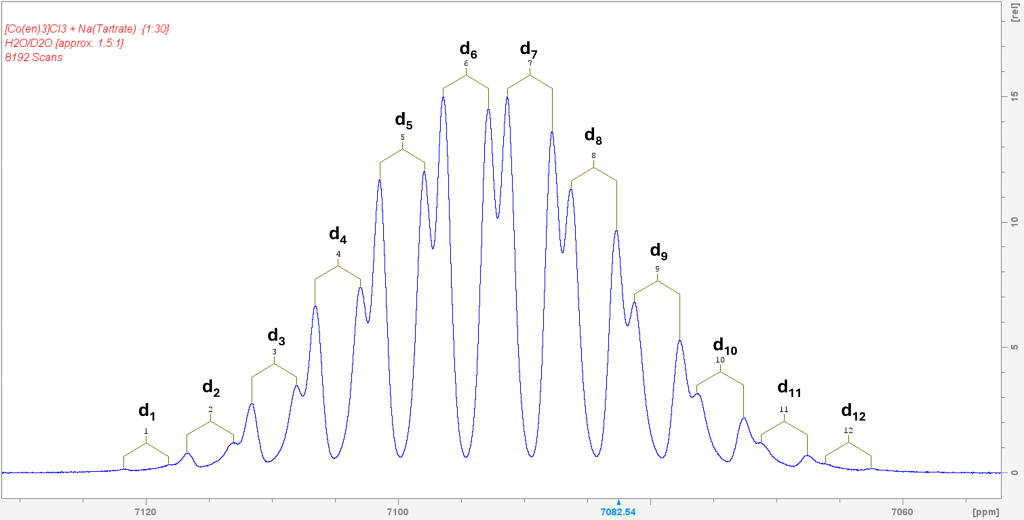
This spectrum was obtained by recording 8192 scans in order to extract the signals of the low-concentration isotopomers d1 and d12 from the noise. The two signals of the isotopomer d0 are missing because its concentration is so low that its signals are lost in the noise. The spectrum shows the isotopomers in equilibrium. Even after 7 days, it remains unchanged. This means that the isotopomer distribution can ultimately be controlled via the H2O/D2O ratio, which is also reported in the literature. To be honest, I was lucky to have hit the H2O/D2O ratio in such a way that the isotopomers d6 and d7 form the maximum.
14N NMR of [Co(en)3]3+
To complete the investigation of [Co(en)3]3+, it seemed sensible to record a nitrogen NMR spectrum. All my attempts to produce a 15N-HMBC spectrum failed miserably. The ligand’s protons produce relatively broad peaks in the 1H NMR spectrum, which probably prevents polarisation transfer to the 15N atoms. I was also unable to find any 15N data in the literature.
The last option was therefore 14N NMR spectroscopy. Fortunately, the appropriate measuring head on our 500 MHz spectrometer had recently been repaired, enabling us to carry out the measurements promptly. Since the signals from primary amines typically appear in the range of 20 to 30 ppm (reference fl. NH3 = 0 ppm) and no dramatic shift was expected due to the coordination of ethylenediamine on cobalt, we searched in a window around this range and were rewarded with a broad signal at -17 ppm.
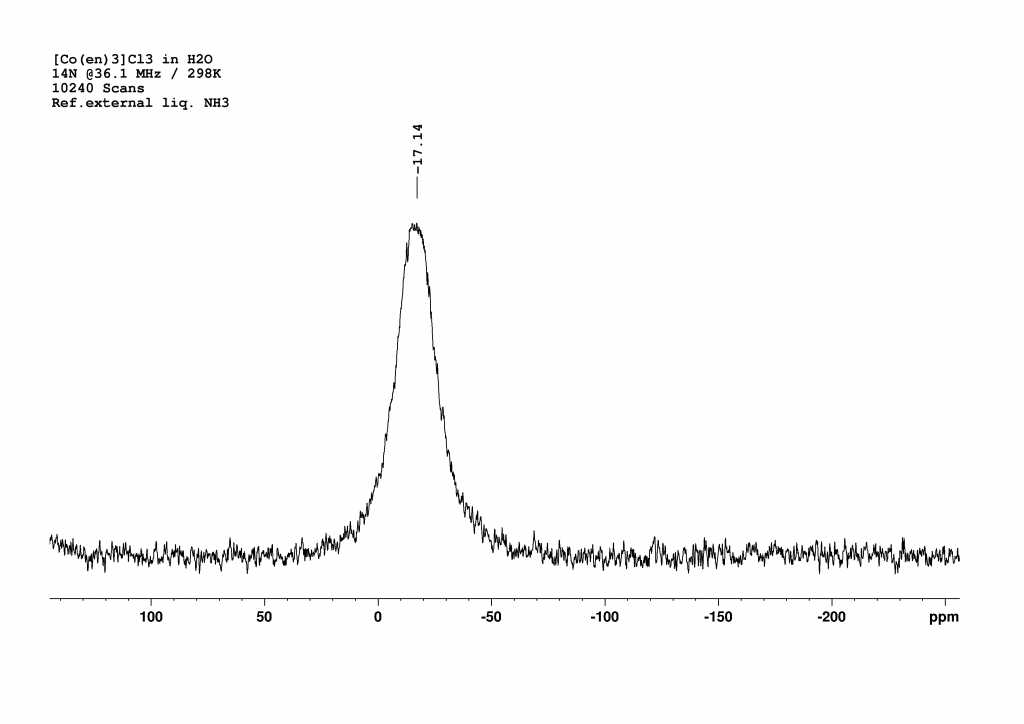
A measurement time of 48 minutes (10240 scans, with a relaxation delay of 0.01 s and an acquisition time of 0.28 s) was necessary to obtain a good spectrum. The peak is very broad (FWHM = 720 Hz), which is unsurprising given that the nitrogen atoms are not symmetrically surrounded. I was somewhat surprised that the nitrogen atoms are more strongly shielded than in the free ligand. I currently have no explanation for this.